La enfermedad de Alzheimer (EA) es una enfermedad neuro-degenerativa de las células cerebrales, las neuronas:
* de carácter progresivo e irreversible,
* de origen todavía desconocido,
* y frente a la que no existe, hoy en día, ningún tratamiento capaz de curarla o prevenirla.

Es la causa de invalidez, dependencia y mortalidad más frecuente en los mayores.
Que es la enfermedad:
El término demencia deriva del latín "demens, dementatus" que significa sin mente. La demencia es definida por la Organización Mundial de la Salud, en la Clasificación Internacional de las enfermedades - Décima Edición (CIE-10, 1992), "como un síndrome debido a una enfermedad del cerebro, generalmente de naturaleza crónica o progresiva, en la que hay déficits de múltiples funciones corticales superiores.... que repercuten en la actividad cotidiana del enfermo".
Entre las funciones corticales superiores que el enfermo va perdiendo figuran la memoria, el entendimiento, el juicio, el habla, el cálculo, el pensamiento, la orientación, etc. No todas se deterioran simultáneamente, sino que es un proceso continuo en el que cada vez se percibe mayor número de funciones afectadas y con progresivo mayor deterioro, siendo generalmente la memoria la primera observación de alteración que percibe el enfermo o sus parientes más próximos. La pérdida única de la memoria sería una amnesia, y el deterioro único de la misma, una dismnesia. En ningún caso, si no existe otra alteración cognoscitiva se puede hablar de demencia.
El diagnóstico de demencia, hoy en día y en la mayoría de los paises, se hace siguiendo las recomendaciones propuestas por Organización Mundial de la Salud (OMS) en la Décima Clasificación Internacional de Enfermedades (CIE-10, 1992) y por la Asociación Americana de Psiquiatría (American Psychiatric Association) -recogido en el Manual de Diagnóstico Estadístico (DSM-IV, 1994) - y son los siguientes:
Según la CIE-10 en la demencia se encuentran estos síntomas (que pueden evaluarse objetivamente con pruebas específicas en muchos casos siempre que sea necesario):
Alteración para registrar, almacenar y recuperar información nueva.
Pérdida de contenidos mnésicos, o memorizados, relativos a la familia o al pasado.
La demencia es más, y más profunda y anómala, que una dismnesia o alteración patológica de la memoria.
Existe reducción en el flujo de ideas
Existe deterioro en el proceso de almacenar información
Dificultad para prestar atención a más de un estímulo a la vez
Dificultad para cambiar el foco de atención:
1.- Deterioro de memoria*
2.- Deterioro del pensamiento y del razonamiento*
3.- Interferencia en la actividad cotidiana
4.- Existe una conciencia clara inicialmente, pero hay la posibilidad de superposición delirio/demencia
*= Demostrados por exploración psicopatológica y testimoniadas por informante.
Cuales son sus diez primeros sintomas:
La enfermedad se manifiesta de forma insidiosa y progresiva, por lo que no siempre es fácil determinar cuando comenzó.
Los primeros síntomas son dificultades de memoria y modificaciones del comportamiento y del humor.
Los 10 primeros síntomas del Alzheimer:
1- Pérdida de memoria
Son olvidos de cita, de fecha, de encargos…siempre de hechos recientes: “Quién vino a verme ayer?”, “Que hemos comido al almuerzo?”, “Quién llamo al teléfono?”
2- Repetición frecuentes de preguntas a pesar de recibir respuestas
“¿Qué hora es?”, “¿Qué hora es?”…o “¿Cuando viene mi madre?,“¿Cuándo viene mi madre?”
3- Colocación de cosas en lugares equívocos
Se encuentran las llaves del coche en la basura, sus gafas metidas en una zapatilla…No se acuerda donde coloco las cosas.
4- Dificultad para recordar el nombre de objetos usuales
“Dame la…la….Ah! no me acuerdo como se llama”
5- Pérdida del sentido de la orientación con respeto al tiempo y o al lugar
“¿Qué día somos?” cuando es obviamente un domingo, “¿Donde estoy?” cuando está en casa. Perderse en el camino de la panaderia donde compra el pan cada mañana
6- Dificultades a realizar gestos simples y familiares
No puede abrir con llave, se equivoca en el manejo del cambio de marchas del coche…
7- Pérdida de interés y de motivación para las actividades que antes se disfrutaba
Deja de leer el periódico, de ver su emisión favorita a la tele, de jugar al mus con sus amigos…
8- Dificultades para realizar tareas fáciles
Se equivoca en la gestión de su cuenta bancaria o tiene difícil hacer un talón. No logra hacer una llamada telefónica
9- Cambios bruscos en el humor
Se pone iracundo, de mal humor… sin razón de ser
10- Dificultad para manejar objetos muy familiares
Se equivoca al utilizar los cubiertos cuando come. Sabe lo que es un peine pero ha olvidado como peinarse.
Cambios de Humor y comportamiento:
Poco a poco, el enfermo disminuye el interés por las cosas que le rodean, se repliega sobre sí mismo y reacciona menos a las emociones, toma menos la iniciativa, se vuelve cada vez más irritable. Ya no es el mismo.
A veces, es un hecho importante el que va a llamar la atención: se pierde en un lugar no familiar u olvida un acontecimiento importante (el aniversario de su boda).
Es ahora cuando las dificultades de memoria, que podían parecer banales, y los cambios en el comportamiento toman su verdadero significado.
Cuales son sus fases:
Se estima que un enfermo tiene un promedio de 10 a 12 años de vida después del diagnostico.
¿Qué va a pasar?
Se describen 3 fases o grados que sirven a los médicos para definir el estado del enfermo en el marco de la evolución: las fases ligera, moderada y severa.
¿Cuál es el interés de estas fases?
Sirven al médico para definir el estado del enfermo en el marco de la evolución de la enfermedad. El diagnostico precisa que es una enfermedad de Alzheimer “probable” en fase ligera o moderada, a veces severa cuando el diagnostico se realiza tarde.
Sirven para seleccionar el o los medicamentos. Por ejemplo, los inhibidores de la colinestera se utilizan a las fases ligeras a moderadas.
Sirven para la evaluación de la pérdida de autonomía. Con una enfermedad de Alzheimer en fase ligera, no se consigue beneficiarse de la Ley de Dependencia o de otras ayudas que se reservan para una fase con dependencia.
 Fases o Etapas
Fases o Etapas
Fase Ligera
– Memoria
El enfermo olvida sus citas, las llamadas telefónicas, el nombre de las personas (relaciones o amigos), los objetos familiares. Tiene dificultades para seguir una conversación, se equivoca en sus cuentas, no paga las facturas.
– Comportamiento
Está sujeto a bruscos cambios de humor. Monta en cólera cuando se percata de que ha perdido el control sobre los elementos que le rodean. Tiene tendencia a aislarse en un entorno familiar que conoce bien: sale menos y no quiere ver a sus amigos.
– Lenguaje y comprensión
Aunque el enfermo continúe razonando y comunicándose bien con los otros, tiene, sin embargo, problemas para encontrar las palabras precisas; sus frases son más cortas; mezcla ideas que no tienen relación directa entre sí.
– Coordinación de gestos espontáneos y movimientos corporales
En esta fase, el enfermo todavía está bien. No se pierde y aún puede conducir, se viste solo y come bien.
– Actividades de la vida diaria
Es capaz de realizarlas sin demasiados problemas, incluso sus actividades profesionales. De hecho, todavía no está afectado más que por pérdida de memoria.
Fase Moderada
– Memoria
La memoria se altera progresivamente.El enfermo olvida los sucesos recientes. No se acuerda de lo que acaba de comer; acusa a sus amigos de abandonarlo porque no vienen a visitarlo. No puede asimilar o comprender los hechos nuevos: un matrimonio o el fallecimiento de un pariente. Sin embargo, el recuerdo de hechos lejanos persiste aunque los sitúe mal en el tiempo en que transcurrieron: el enfermo pide noticias de su madre fallecida recientemente o menciona a personas a las que no ha visto desde hace años.
– Comportamiento
Este es el momento de las reacciones agresivas, desproporcionadas respecto al motivo que las ha desencadenado. Puede acusarle a Vd. de robarle si no encuentra su monedero; grita e incluso se vuelve agresivo si se insiste para que se bañe. Cuanto más depende de otros más se irrita. Su fatiga aumenta y no hace nada sin que se le estimule.Experimenta miedos injustificados; un ruido, una cortina que se mueve o una luz pueden desencadenarlos.Camina durante horas de un lado a otro. Se levanta durante la noche y prepara su maleta para volver a casa
– Lenguaje y comprensión
El conjunto de la comunicación con los demás se hace más difícil: habla menos, su vocabulario se empobrece, repite siempre las mismas palabras o las mismas frases durante horas. Cuando responde a las preguntas lo hace lentamente, buscando las palabras; no acaba las frases.
– Coordinación de gestos
Sus gestos son imprecisos: se abrocha mal los botones, sostiene mal su tenedor o su cuchillo. Pierde el equilibrio. Se golpea con facilidad y las caídas son frecuentes. Se mueve lentamente y necesita que lo ayuden para ir a su habitación o al baño. Pueden aparecer movimientos anormales como temblores, contracturas musculares o convulsiones.
– Actividades de la vida diaria
Su creciente confusión hace que le resulte cada vez más difícil enfrentarse a la vida diaria. No es capaz de elegir:
- entre sus ropas, pues se viste sin importarle como, y sin tener en cuenta la estación o los convencionalismos sociales;
- entre los platos que ponen en la mesa;
- entre las etapas habituales de su baño o ducha: ¿cuándo quitarse la ropa?,¿cuándo enjabonarse?, ¿cuándo secarse?
Por otro lado pierde su autonomía ya que no puede conducir, ni viajar en metro o en autobús sin compañía. Se pierde incluso en un trayecto que le es familiar.
Puede dedicarse a actividades peligrosas para si mismo y para los demás como abrir la llave del gas sin encenderlo, u olvidar su cigarrillo y prender fuego por accidente.
Es decir, el enfermo ha de ser vigilado las 24 horas del día, lo que significa que sus familiares deben prestarle atención constante.
Fase Severa
– Memoria
El enfermo olvida los hechos recientes y pasados. No reconoce a su cónyuge o a sus hijos. Sin embargo, conserva la memoria emocional. Se da cuenta de la persona que le cuida, le ayuda y le quiere. Este hecho debe estar siempre presente en la mente de quien se ocupa de él.
– Comportamiento
Su humor es imprevisible: grita, llora, se agita. No reacciona coherentemente ante una situación, ni comprende una explicación.
– Lenguaje y comprensión
El enfermo balbucea, repite palabras sin pies ni cabeza, y solo utiliza correctamente algunas palabras concretas. No comprende lo que se le dice.
– Coordinación de los gestos
No controla sus gestos. No sabe levantarse, sentarse o andar. Le cuesta trabajo tragar. No controla los esfínteres y aparece incontinencia.
– Actividades de la vida diariaHan desaparecido totalmente. Con gran frecuencia permanece en la cama, lo que conduce a la aparición de llagas en los puntos de presión e infecciones respiratorias.
La muerte sobreviene generalmente debido a una enfermedad asociada (cáncer, accidente cardíaco o vascular cerebral), a veces como consecuencia de permanecer encamado (infecciones de las llagas, infecciones respiratorias y o urinarias…).
Cuales son sus causas:
La causa (o las causas) de la enfermedad de Alzheimer es por el momento desconocida, pese a los considerables esfuerzos realizados por numerosos equipos de investigación.
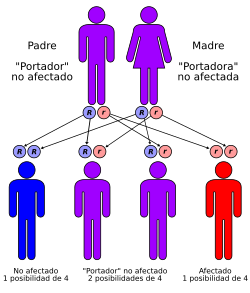
Las formas “familiares” de la enfermedad
Si, se conocen formas de la enfermedad que tienen un origen genético bien probado: son las formas “familiares”. Son muy raras (menos de 1% de los casos) y aparecen siempre en sujetos de menos de 65 años, pudiendo darse incluso en sujetos muy jóvenes (25-30 años).
Se deben a mutaciones anormales en ciertos cromosomas y el portador de la mutación transmitirá la enfermedad a uno de cada dos hijos, sea cual sea su sexo. Se llama “transmisión autonómica dominante”.
Las mutaciones afectan a genes situados en tres cromosomas diferentes, que codifican para proteínas que intervienen en el metabolismo del péptido amiloïde, elemento fundamental de las placas seniles (precursores de las proteínas amiloide, presenilina 1 y 2, situadas respectivamente en los cromosomas 21, 14 y 1).
Las formas “esporádicas”
En el 99 % de los casos, son formas “esporádicas” es decir que aparecen sin que haya una relación directa con las anomalías genéticas que acabamos de mencionar.
En esto casos, que son la inmensa mayoría, desconocemos la causa o las causas de la enfermedad, a pesar de los esfuerzos de la investigación. No obstante, se conocen mejor determinados factores que desempeñan un papel importante en su aparición, y que llamamos “factores de riesgo”.
¿Qué es un factor de riesgo?
Es importante comprender que un factor de riesgo es aquel cuya presencia aumenta las probabilidades de estar afectado por la enfermedad, pero los factores de riesgo no determinan el desarrollo de la misma. Puede Vd. presentar un factor de riesgo y no desarrollar nunca la enfermedad. Por el contrario, puede Vd. desarrollar la enfermedad en ausencia de todo factor de riesgo.
Factores de riesgo genéticos.
Factores otros factores de riesgo.
Factores de proteción.
Cual es su tratamiento:
El tratamiento “farmacológico” se refiere al uso de medicamentos por oposición al tratamiento “no farmacológico” en el cuál las intervenciones terapéuticas utilizan técnicas (por ejemplo, psicoestimulación sin recurrir a medicamentos).
“Cuando veo el cajón de la mesilla de mi padre, estoy aterrada. Hay de todo. Medicamentos que toma desde hace más de 20 años, gotas para su tensión, pastillas para aliviar sus piernas…y otros que desconozco."
"Nos llamo la atención cuando empezó a tener problemas con su cabeza. Utilizaba sus pastillas al azar. Cuando supimos que tenía Alzheimer, el médico nos dijo que había que limpiar el cajón y asegurarse bien que tomaba el tratamiento a los horas previstas, que tragaba los comprimidos de verdad…en fin, nos encargo de vigilar su tratamiento” (testimonio de una hija que cuida a su padre).
Hace unos años, este capítulo no existía porque no había medicamentos específicos para tratar la enfermedad de Alzheimer.
Ahora, existen medicamentos cuya finalidad es retrasar la evolución de sus síntomas.
Pero, como no se sabe cuál es la causa (o las causas) de le enfermedad, no existe, hoy en día, un “medicamento” capaz de curarla o de prevenirla.